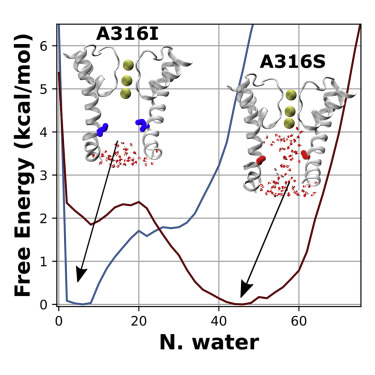
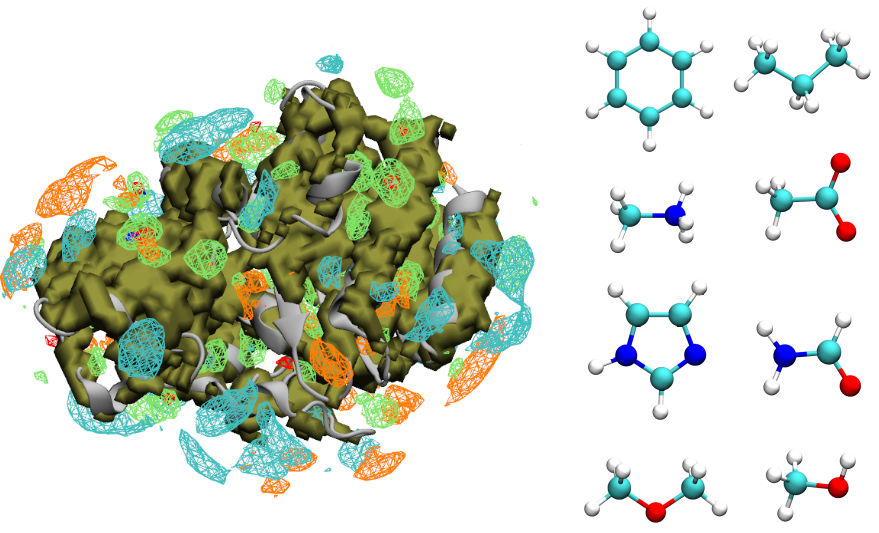
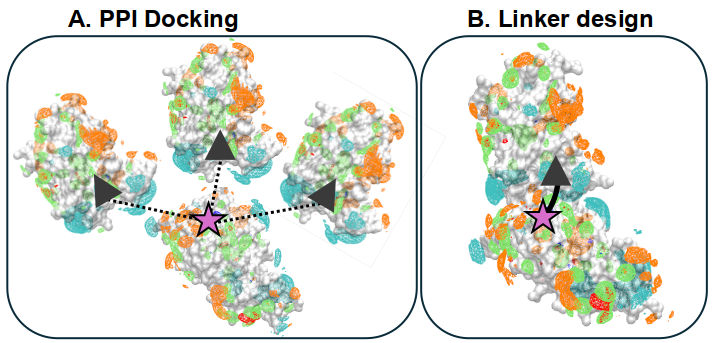
Biophysics and Machine Learning
It is often useful to leverage computational biophysical data and machine learning to fill gaps in experiments. I’ve used this approach with the chaperone Hsp70, BK channel, and to predict druggable sites. 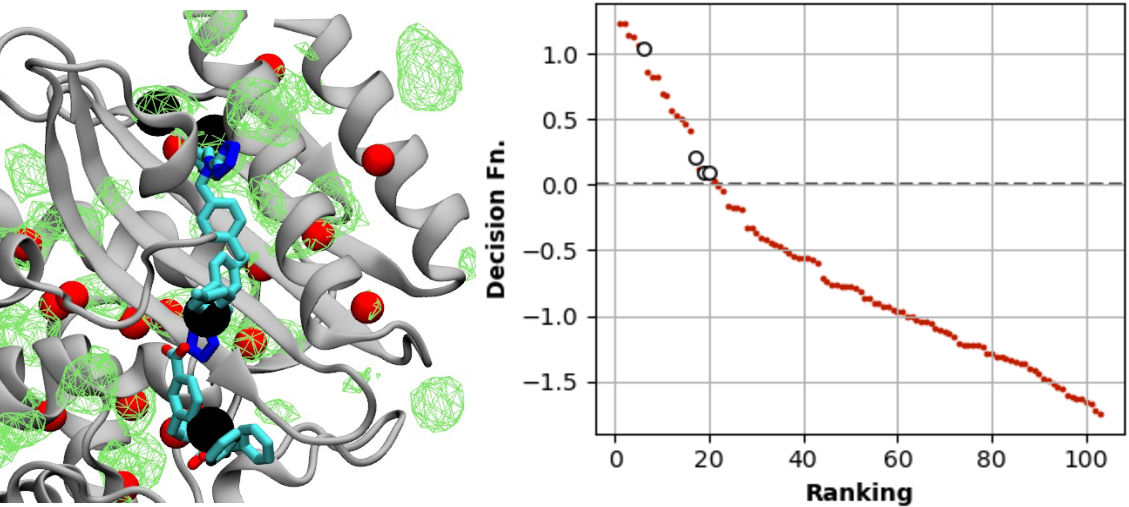
Read more