
Computer-aided drug discovery
The MacKerell lab has developed a computer-aided drug discovery method called Site Identification by Ligand Competitive Saturation (SILCS), which uses convential molecular dynamics simulations of a target protein solvated by a saturated solution of water and small molecule fragments. The result is a comprehensive 3D free energy affinity map of each fragment (FragMap) across the complete surface of the protein as a dynamic, rather than static object. The solutes and FragMaps are depicted below. A key breakthrough to this technology was the use of an enhanced type of molecular simulation technique, oscillating-chemical potential Grand Canonical Monte Carlo simulations, to enhance the sampling of small molecule fragments within hard-to-reach binding pockets. This approach is somewhat computationally-analogous to the experimental method of Fragment-based screening using crystallography or NMR, but is much faster than both and improves on the lack of protein flexibility in crystallography and the spatial resolution of NMR.
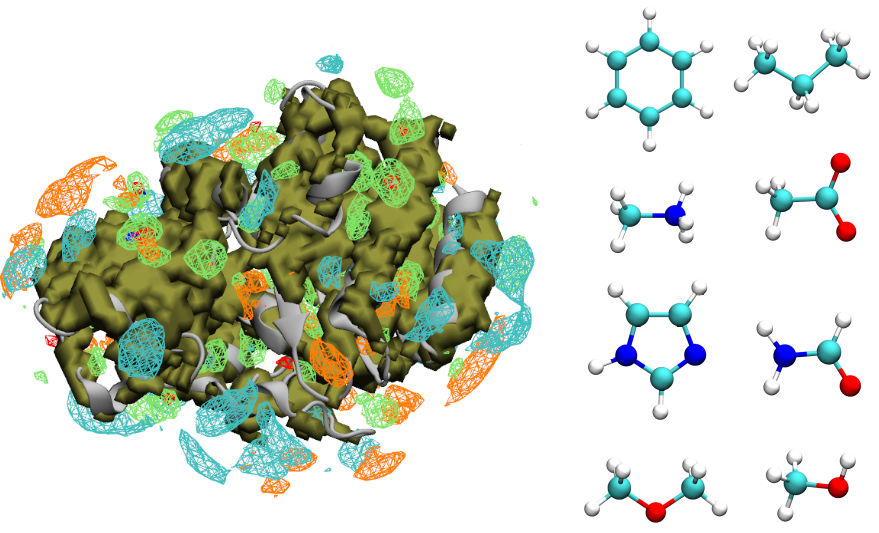 You can read more about the method of GCMC-MD at the MacKerell lab's Research page, or by reading this paper. There is a fantastic-looking and easy-to-use Interactive demo on the SilcsBio (R) website for a hands-on and visual guide of the method.
You can read more about the method of GCMC-MD at the MacKerell lab's Research page, or by reading this paper. There is a fantastic-looking and easy-to-use Interactive demo on the SilcsBio (R) website for a hands-on and visual guide of the method.
Disclaimer: The SILCS GCMC/MD simulation method and related tools is the basis of a private company, SilcsBio, LLC, of which my advisor Alexander D. MacKerell, Jr. is co-founder and CSO. I am working for Alex as part of the University of Maryland Balitmore and the NIH (T32 grant), not SilcsBio; however, we are all (UMB, NIH, SilcsBio) invested in essentially the same goals, the University of Maryland actively exchange between industry and academia, and SilcsBio, LLC has a free academic license.
You can read more about the method of GCMC-MD at the MacKerell lab's Research page, or by reading this paper. There is a fantastic-looking and easy-to-use Interactive demo on the SilcsBio (R) website for a hands-on and visual guide of the method. Disclaimer: The SILCS GCMC/MD simulation method and related tools is the basis of a private company, SilcsBio, LLC, of which my advisor Alexander D. MacKerell, Jr. is co-founder and CSO. I am working for Alex as part of the University of Maryland Balitmore and the NIH (T32 grant), not SilcsBio; however, we are all (UMB, NIH, SilcsBio) invested in essentially the same goals, the University of Maryland actively exchange between industry and academia, and SilcsBio, LLC has a free academic license.