Biophysics and Machine Learning
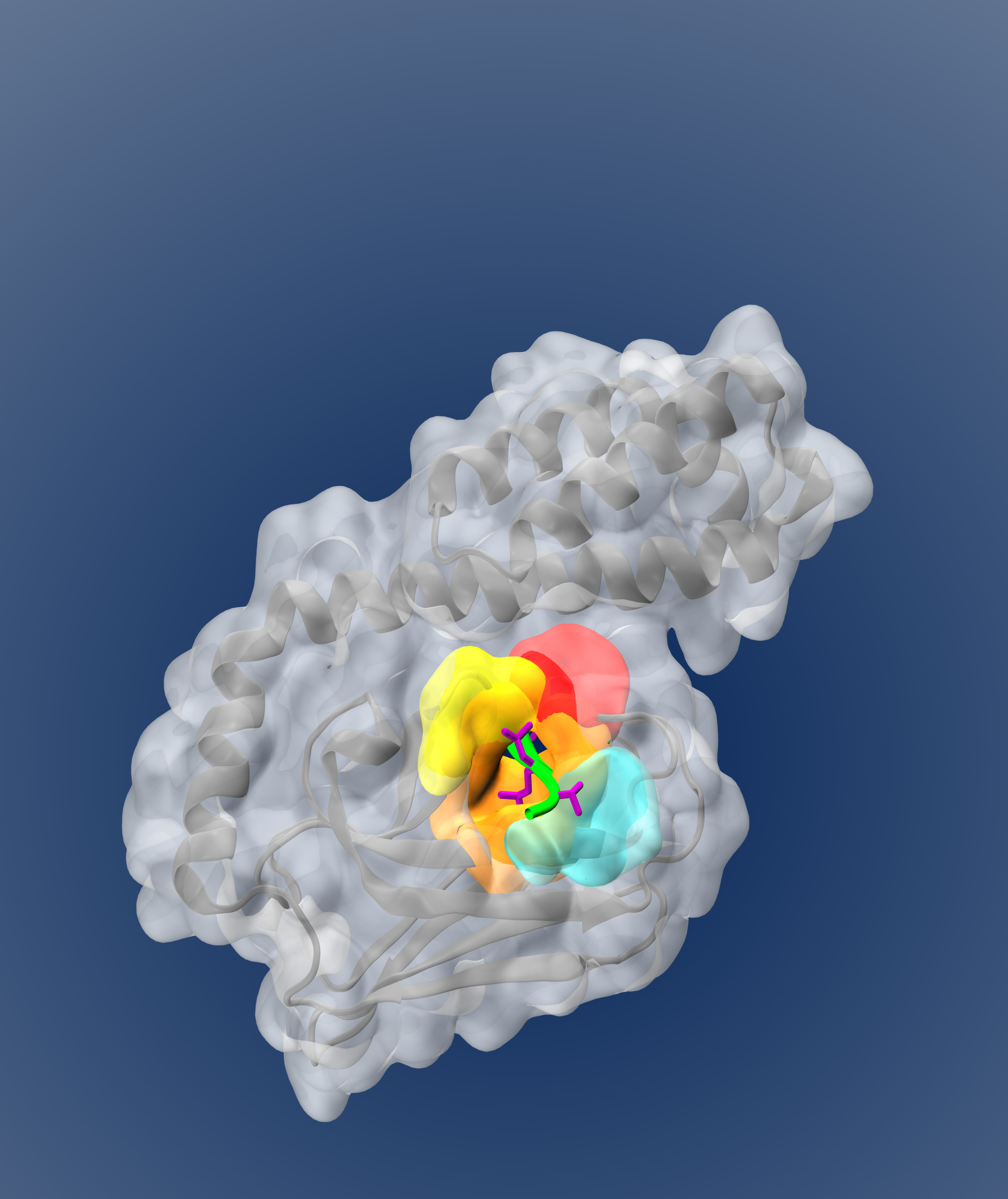
Predicting druggable binding sites on proteins (SILCS-Hotspots)
SILCS provides a powerful physics-based set of data which can be leveraged to predict novel crytpic and allosteric binding sites.Identifying druggable sites on target proteins is a critical first step in a computer-aided drug design (CADD) campaign. We used the SILCS technique (GCMC/MD) to sample the binding affinity of various co-solutes at all regions of the protein. Proteins are inherently dynamic objects, so to accurately identify ligand binding sites including cryptic sites and allosteric sites, accounting for protein flexibility as in SILCS simulations is critically important. We analyzed the distribution of fragment binding Hotspots in relation to the binding sites of crystallographic ligands across a training set of several proteins, then extracted features to be used in a SVM model. The model could be predict the binding sites of proteins in an independent validation set of proteins, and even predict novel druggable sites. You can check out the paper here, and can utilize the code via a free academic license to SILCS at silcsbio.com.
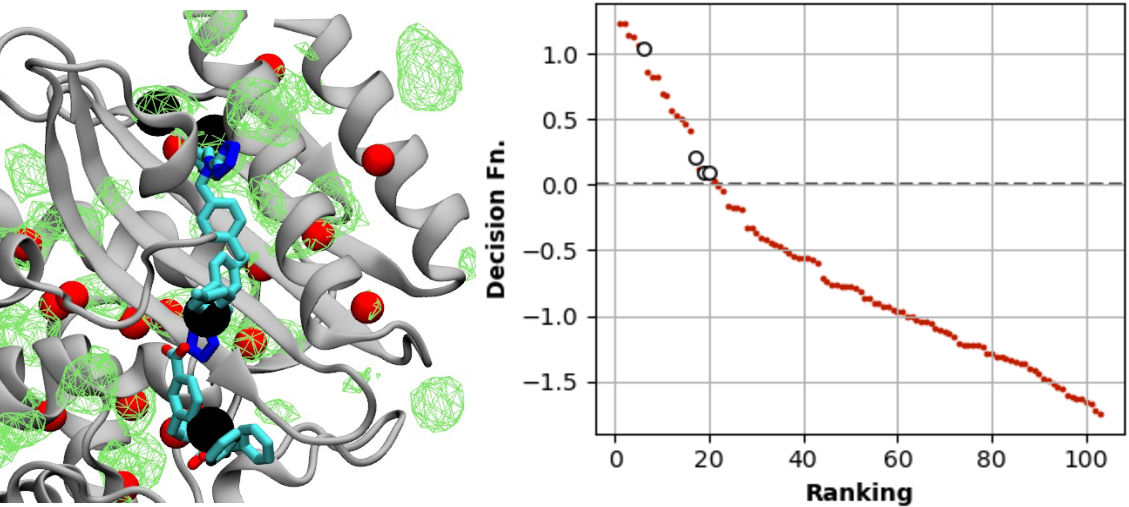
BK Channel voltage gating, predicting effect of mutations
Some mutations to the BK channel can have incredibly sensitive effect on function, yet many have minimal impact.The BK channel is a critical but challenging drug target due to its importance in the CNS and the heart. When we began to build this model, we were motivated by the fact that high-resolution structures of the channel in active and inactive states was available, and that there were functional data for many (>450 mutations) available. Still, that was only ~2-3% of all possible single mutations, so we constructed a physics-based description of the effect of each possible mutation using MD simulations and computational mutagenesis, then trained these descriptors on the mutagenesis data set using machine learning methods. We validated our predictions by testing four mutantions our collaborator Jianmin Cui's lab, and saw remarkable agreement suggesting that these mutations lie at a site critical for voltage gating. You can check out the paper here or the code on GitHub.
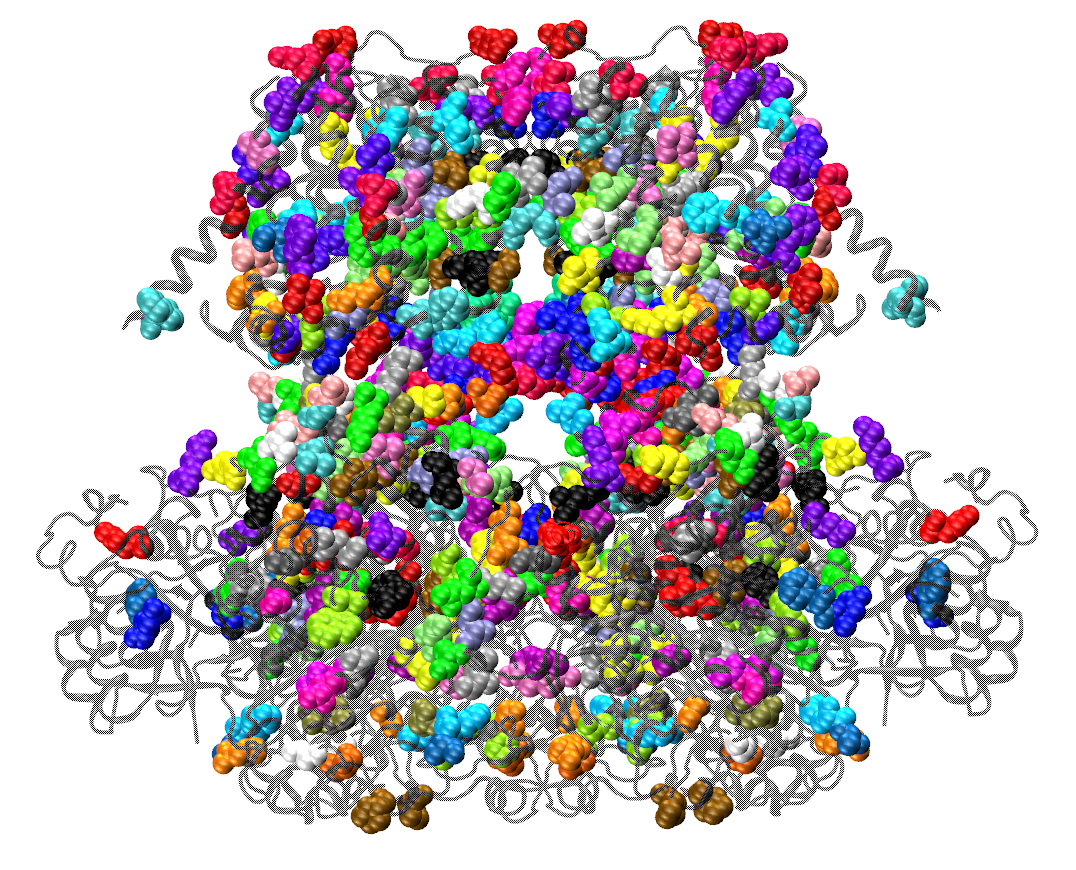
Hsp70 molecular chaperone, peptide recognition
Molecular chaperones like DnaK help fold misfolded proteins, but how do they distinguish well-folded, misfolded, and disordered regions?Hsp70s part of a special group of proteins, molecular chaperones, that help perform many roles in cellular quality control. They do this in part thanks to a remarkable ability to bind to a wide variety of misfolded proteins without binding to well-folded or intrinsically-disordered proteins. The physical basis for the molecular chaperone Hsp70’s promiscuous selectivity for many substrate peptide sequences, but not all, is challenging to understand and predict. We collaborated with the group of chaperone expert Lila Gierasch at UMass Amherst to ensure the model was designed with the best experimental guidance. Structural analysis directly informed an MD simulation protocol to derive an MD "basis set", which we trained to reproduce low-resolution binding fluorescence experiments. The model performed quite well on higher-reoslution x-ray based data, including predicing the preferred orientation of peptides with state of the art accuracy. You can read the paper here, or check out the code on Github.